Il villaggio globale in cui va sempre più trasformandosi il mondo moderno ha portato all’emergenza, nei paesi europei industrializzati di patologie da noi rare o addirittura scomparse. E’ il caso di alcune malattie infettive come la difterite e la tubercolosi e di alcune emoglobinopatie frequenti nei territori con malaria endemica, come l’anemia falciforme e l’emoglobinosi C, che rappresenta un fattore di protezione dall’infestazione da parte del Plasmodium Malariae.
L’emoglobinosi C è dovuta alla presenza di una una variante dell’emoglobina, dovuta a una mutazione nel gene della beta globina, che causa la sostituzione di un acido glutammico con una lisina in posizione 6 nella catena globinica. Un’altra sostituzione aminoacidica, che si manifesta nello stesso sito della beta globina, causa l’emoglobinopatia a cellule falciformi (HbS).
Tale emoglobina precipita all’interno degli eritrociti, provocando la formazione di cristalli che rendono le emazie meno deformabili con iperviscosità del sangue. La riduzione dell’emoglobina, accentuata da ipossemia, ne favorisce la precipitazione con peggioramento dell’iperviscosità. Le emazie contenenti i cristalli vengono rimosse dalla milza. E’ dimostrata anche una alterazione della membrana degli eritrociti, dovuta ad una diversa composizione fosfolipidica.
La malattia viene trasmessa geneticamente con un meccanismo autosomico recessivo; la percentuale di HbC è quasi 100% nei soggetti omozigoti, mentre negli eterozigoti può arrivare al 35% in rapporto con la penetranza del gene anomalo. Può esserci associazione con malattia da HbS (con peggioramento della falcizzazione delle emazie) o con beta talassemia.
La prevalenza della HbC è addirittura del 40-50% nell’Africa Occidentale (Burkina Faso, Costa d’Avorio, Ghana). La malattia si riscontra anche nel Togo e nel Benin (20%), nelle persone di discendenza africana nei Caraibi (3,5%) e negli USA (3%), nel Nord Africa (da 1 a 10% in Marocco e Algeria) e nel Sud Europa (Italia, Turchia). I soggetti eterozigoti per l’HbC (AC) sono asintomatici e possono presentare lieve microcitosi e aumento della resistenza degli eritrociti all’emolisi. Gli omozigoti per HbC (CC), di solito, compensano l’emolisi con la splenomegalia. Presentano un aumento del rischio di ipersplenismo, litiasi biliare, deficit di folati e aggravamento dell’anemia dopo infezione con Parvovirus B19.
Frequentemente la storia clinica è silente, trattandosi di emolisi cronica compensata. Possono essere presenti dolori muscolari o articolari o articolari, colelitiasi da calcoli di bilirubina (pigmentati), splenomegalia con eventuali citopenie da ipersplenismo, anomalie mascellari e mandibolari diagnosticabili radigraficamente dovute a iperplasia del midollo emopoietico.
In genere l’anemia è modesta o assente, sono presenti in misura variabile segni di iperemolisi (reticolocitosi, incremento dell LDH e della bilirubina indiretta, riduzione dell’aptoglobina).
Lo striscio periferico dimostra la presenza di cellule bersaglio, stomatociti, corpi inclusi citoplasmatici
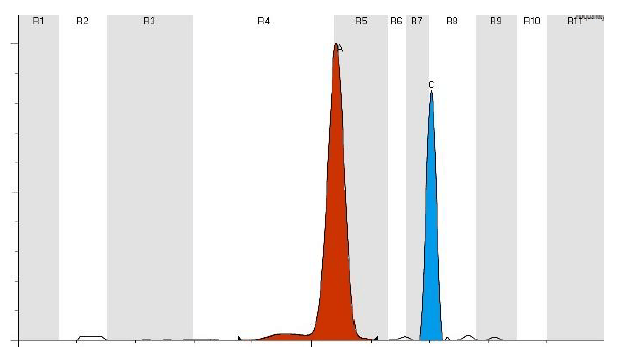
La malattia è diagnosticata con l’elettroforesi dell’emoglobina. Questo permette di separare l’emoglobina in base alle proprietà di carica elettrica della globina. Mutazioni sulla catena della molecola introducono modifiche strutturali e di carica per cui si hanno differenze per cui si hanno differenze di migrazione che permettono di separare le catene anomale. L’emoglobina C presenta una banda intensa a livello HbA2.
Medical Systems a tal fine offre ed utilizza per la determinazione e l’dentificazione di tutte le emoglobine sia strumentazioni in gel d’agarosio (SAS1-SAS2 e/o SAS3-SAS4) ed una strumentazione in tecnica capillare con metodo di Isoelettrofocalizzazione denominato V8 Nexus.
In merito alla strumentazione in gel d’agarosio (SAS1-SAS2 e/o SAS3-SAS4), il protocollo per l’elettroforesi dell’emoglobina comprende l’uso di sue sistemi utilizzati in due fasi. L’elettroforesi iniziale fatta con tamponi alcalini ( kit COMBI HB ALCALINA cod. 05750345). Tuttavia, a causa della somiglianza elettroforetica di molte emoglobine, tra cui l’HbC, la valutazione deve essere integrata da elettroforesi con tampone acido (kit COMBI HB ACIDA cod. 05750347) al fine di misurare una proprietà diversa dalla carica.
Con lo strumento in tecnica capillare V8 Nexus la separazione e la quantizzazione delle emoglobine avviene mediante Focalizzazione isoelettrica capillare con l’utilizzo del kit V8 V-CE Haemoglobin IEF cod. 800900.
Questa separazione IEF utilizza i diversi punti isoelettrici di ciascuna delle molecole di emoglobina se separata in un gradiente di pH.
La tecnica sfrutta il fatto che la corsa di un analita cambia in base al pH di ciò che lo circonda. Pertanto, una specie che si trova in una regione di pH al di sotto del suo punto isoelettrico sarà caricata positivamente e migrerà verso il catodo. Mentre migra attraverso un gradiente di pH in aumento, tuttavia la carica complessiva della proteina diminuisce finchè non raggiunge la regione di pH che corrisponde al suo punto isoelettrico. A questo punto non ha nessuna carica e quindi la migrazione cessa.
Di conseguenza, gli analiti vengono focalizzati in bande fisse e nette, ciascuno posizionato ad un determinato e preciso punto di gradiente di pH corrispondente al suo punto isoelettrico.
Poiché come detto in precedenza della somiglianza elettroforetica delle molte varianti emoglobiniche per struttura e carica, l’elettroforesi capillare V8 Nexus con il metodo di isoelettrofocalizzazione permette una risoluzione estremamente ottimale.