L’aptoglobina umana (Hp) è una α2-sialoglicoproteina che elettroforeticamente migra nella regione delle α2 globuline. Sintetizzata per la maggior parte dagli epatociti, si trova nel plasma alla concentrazione di 0,45-3,0 mg/ml. Si tratta di una proteina tetramerica (αβ)2 costituita da due catene polipeptidiche α leggere e da due catene β pesanti covalentemente associate da legami disolfurici intracatena in una struttura quaternaria. L’aptoglobina è stata molto studiata sotto il profilo genetico a causa dell’esistenza di differenti fenotipi ampiamente rappresentati nella popolazione umana. Il polimorfismo genetico è caratterizzato da tre differenti fenotipi strutturali : Hp1-1, Hp2-2 e Hp2-1 che sono determinati dalla espressione combinata da una coppia di alleli autosomici codominanti Hp1 e Hp2 del gene Hp.La catena α esiste nei due comuni fenotipi α1 e α2 ed è responsabile dell’osservato polimorfismo genetico. La catena β, essenzialmente non polimorfa, è praticamente identica in tutti e tre gli alleli; soltanto poche rare varianti sono state descritte. il polimorfismo genetico dell’aptoglobina è generalmente associato alla predisposizione nonché alla evoluzione di numerose patologie.
Hp2-2, ad esempio, è sovra rappresentato nelle malattie autoimmuni e si ritiene costituisca il maggior fattore di rischio in numerose patologie correlate allo stress ossidativo. Nei pazienti diabetici, l’omozigosità Hp2 incrementa la probabilità dello sviluppo di nefropatia diabetica e di retinopatia. Inoltre l’associazione tra Hp2-2 e lesioni aterosclerotiche conduce all’ipertensione e a malattie cardiovascolari.
Hp2-1, a sua volta, è strettamente correlato con numerose forme di cancro.
La concentrazione di aptoglobina plasmatica varia in rapporto all’età e a numerose condizioni patologiche . praticamente assente nel corso della vita fetale e neonatale, la sua concentrazione aumenta dalla nascita fino all’età adulta per portarsi ad un valore normale intorno 100-300 mg/dl.
Nel corso di processi infiammatori, in presenza di infezioni, traumi, ustioni, collagenopatie, malattie cardivascolari, in particolare nell’infarto del miocardio, nelle neoplasie metastatizzanti si registra un anomalo rapido aumento della concentrazione plasmatica dell’aptoglobina. Viceversa una diminuzione della concentrazione si registra per iperconsumo in tutti i casi di emolisi intra ed extravascolare, eritropoiesi inefficace e in caso di insufficienza epatica.
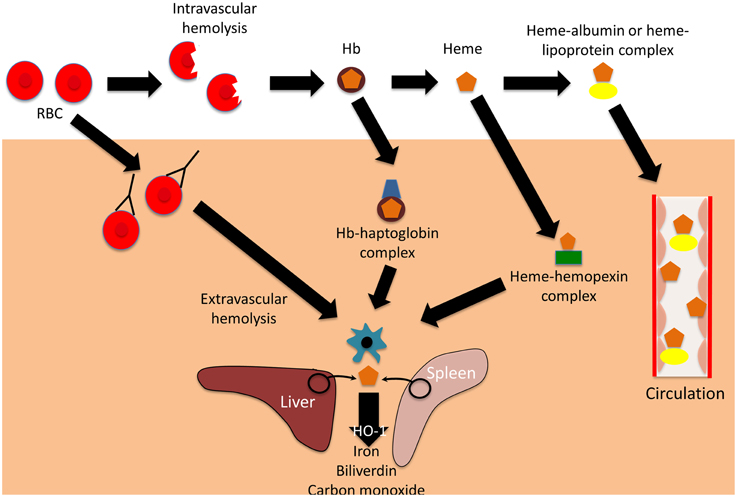
Primariamente sintetizzata dagli epatociti, l’aptoglobina, una volta rilasciata nel plasma, ha una vita media di 2-4 giorni ed è caratterizzata da una elevata affinità sia per l’emoglobina (Hb) derivante dagli eritrociti senescenti in circolazione per emolisi extravascolare, sia per l’emoglobina derivante dalla rottura degli eritrociti per emolisi intravascolare.
Conseguentemente, l’aptoglobina, in presenza di emoglobina, immediatamente ad essa si lega in maniera irreversibile ed il complesso (Hp-Hb), originatosi a seguito delle interazioni molecolari è molto stabile anche sotto varie condizioni di temperatura, di PH e di forza ionica. Il complesso Hp-Hb ha una emivita piuttosto breve (10-30 min) in quanto viene rapidamente captato e rimosso dalla circolazione delle cellule del sistema reticolo endoteliale (SRE).
Il principio di rimozione ha inizio a seguito di una forte interazione che si stabilisce tra Hp-Hb e il recettore proteico CD163 espresso esclusivamente sulla superficie dei monociti e dei macrofagi. L’elevata affinità ligando recettore consente a CD163 di operare da decontaminante dell’emoglobina, in quanto media l’endocitosi dei complessi Hp-Hb nei macrofagi. Singolarmente , sia Hp che Hb non manifestano affinità per CD163.
Nei macrofagi il complesso Hp-Hb è catabolizzato in globina ed eme, quest’ultimo, a sua volta, è degradato dall’eme-ossigenasi. Il ferro rilasciato viene sequestrato dalla ferritina e conservato nelle cellule oppure trasportato nel midollo osseo tramite la transferrina plasmatica per la sintesi di nuova emoglobina. L’aptoglobina non è riciclata.
Il processo appena descritto è molto importante in quanto impedisce che, a seguito di emolisi intravascolare, si determini un possibile danno renale come conseguenza della escrezione nelle urine di emoglobina. Se fosse consentito all’emoglobina di passare attraverso i glomeruli o i tubuli renali si avrebbe non soltanto la comparsa di emoglobinuria, ma si registrerebbe anche un consistente danno ossidativo dovuto al fatto che il ferro presente nell’emoglobina ha la capacità di catalizzare la formazione e l’accumulo di pericolosi radicali ossidrili attraverso una reazione con il perossido di idrogeno.
Qualora, in seguito ad una crisi emolitica, si abbia una liberazione eccessiva di Hb, nel plasma, si registra un rapido consumo dell’aptoglobina plasmatica e la rimozione dei complessi Hp-Hb ad opera dell’SRE che porta in qualche ora ad una condizione di anaptoglobinemia che, in generale persiste per diversi giorni.
Inoltre va sottolineato come l’aptoglobina, concorrendo all’omeostasi del ferro, manifesta attività antibatterica in quanto impedisce ai batteri di utilizzare questo elemento essenziale per la loro crescita.
Interpretazione dei valori
Una riduzione dell’aptoglobina è in grado di supportare una diagnosi di anemia emolitica, specialmente quando è associata ad una serie di fattori: riduzione del numero di globuli rossi circolanti, emoglobina, ematocrito ed un aumento dei reticolociti. Se la conta dei reticolociti fornisce un risultato elevato, ma il livello di aptoglobina è normale, si possono verificare tre casi:
- la distruzione cellulare si sta verificando a livello della milza e del fegato;
- l’emolisi può essere indotta da farmaci che sono stati assunti;
- può essere il segno della presenza di una displasia a carico dei globuli rossi.
Se si riscontrano i sintomi classici di anemia, ma sia la conta dei reticolociti che il livello aptoglobina circolante sono normali, l’anemia è molto probabilmente non è causata dall’emolisi, ma probabilmente si soffre di qualche altro errore nella produzione dei globuli rossi.